
¿Qué es el Síndrome de Behçet?
El Síndrome de Behçet es una enfermedad infrecuente, crónica y de causa autoinmune que provoca inflamación en los vasos sanguíneos de todo el cuerpo. Es una condición compleja, ya que puede afectar varias partes del organismo, como la piel, la boca, los ojos, los genitales, las articulaciones, el sistema digestivo y el sistema nervioso.
Esta enfermedad aparece en forma de brotes (periodos de síntomas activos) y remisiones (periodos sin síntomas), y puede variar mucho de una persona a otra.
¿Cuáles son los síntomas?
Los síntomas del Síndrome de Behçet pueden ser muy variados, pero los más comunes incluyen:
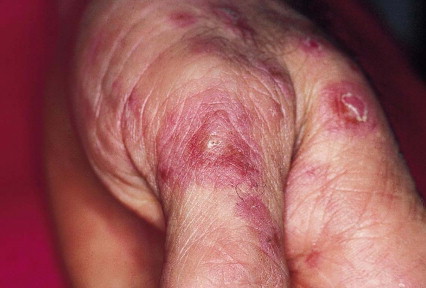
- Úlceras bucales dolorosas (parecidas a aftas), que aparecen de forma repetida.
- Úlceras genitales.
- Inflamación de los ojos (uveítis), que puede causar enrojecimiento, dolor y visión borrosa.
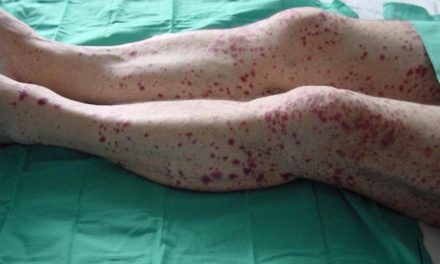
- Lesiones en la piel, como nódulos rojos dolorosos (eritema nodoso) o lesiones parecidas al acné.
- Dolor e hinchazón en las articulaciones (artritis).
- Problemas digestivos (diarrea, dolor abdominal).
- Afectación del sistema nervioso (dolores de cabeza, confusión, dificultad para moverse o hablar).
- Inflamación de vasos sanguíneos que puede causar coágulos o afectar órganos vitales.
¿Cuáles son las causas?
La causa exacta del Síndrome de Behçet no se conoce. Se clasifica hoy como una enfermedad autoinflamatoria sistémica, con componentes autoinmunes secundarios en algunos pacientes. Factores que pueden influir:
- Predisposición genética: algunas personas tienen genes que las hacen más propensas.
- Factores ambientales o infecciones que podrían activar la enfermedad en personas predispuestas.
Es más común en personas de origen mediterráneo, del Medio Oriente y de Asia, y suele comenzar entre los 20 y 40 años.
¿Cómo se diagnostica?
No existe una prueba específica para diagnosticar el Síndrome de Behçet. El diagnóstico se basa en los síntomas clínicos y en descartar otras enfermedades.
El médico puede solicitar:
- Análisis de sangre y orina.Tiene asociación genética con HLA-B51, frecuente en enfermedades autoinflamatorias.Hay ausencia de autoanticuerpos específicos, típicos de enfermedades autoinmunes clásicas.
- Exámenes oculares.
- Estudios de imagen si hay afectación neurológica o vascular.
- Biopsias de piel o úlceras en algunos casos.
El diagnóstico puede ser tardío, ya que los síntomas pueden aparecer de manera gradual.
Tratamiento
El tratamiento del Síndrome de Behçet se adapta según los órganos afectados y la intensidad de los síntomas. Aunque no tiene cura, el tratamiento ayuda a controlar los brotes y prevenir complicaciones.
Opciones comunes incluyen:
- Corticoides para reducir la inflamación.
- Inmunosupresores (como azatioprina, ciclofosfamida) para controlar la respuesta del sistema inmunológico.
- Medicamentos biológicos en casos más graves o resistentes.
- Tratamientos locales para las úlceras bucales y genitales.
- Colirio o medicamentos específicos para los ojos, si hay inflamación ocular.
Recomendaciones para los pacientes
- Siga el tratamiento y las indicaciones médicas al pie de la letra.
- Mantenga un registro de los síntomas para identificar brotes y ayudar al médico a ajustar el tratamiento.
- Evite el estrés, ya que puede desencadenar o empeorar los brotes.
- Evite infecciones y controle otras enfermedades que puedan afectar el sistema inmunológico.
- Asista regularmente a sus controles médicos, especialmente si tiene afectación ocular o neurológica.
- Cuide su salud mental: vivir con una enfermedad crónica puede ser difícil, por lo que es útil buscar apoyo emocional o psicológico.
Importante: Aunque el Síndrome de Behçet puede ser impredecible, con un tratamiento adecuado y seguimiento médico constante, muchas personas logran llevar una vida activa y plena.


{kind=link}